Back To Protocol
RNA-based therapeutics have been under development for over 30 years however, the COVID-19 pandemic accelerated their development, chiefly with mRNA-based vaccines against the SARS-CoV2 virus. A key part of the technology are the lipid nanoparticles (LNPs) used to deliver RNA. Naked RNA cannot be injected as it is immunogenic, susceptible to enzymatic degradation, and is not taken up by cells. RNA packaged in LNPs protects it from degradation while circulating and allows it to enter cells and release the contents into the cytoplasm so the RNA can be used by ribosomes for protein synthesis.
Protocol
This protocol is designed with predetermined molar percentages of lipids and RNA:lipid ratio. Other percentages and ratios may be prepared but calculations for alternative formulations should be adjusted accordingly by the end user.
Preparation of the lipid mixture
This procedure is for a 25 mM lipid stock of 50:10:38.5:1.5 % ionizable lipid:DSPC:cholesterol:PEG.
- Bring all lipids to room temperature.
- Bring ionizable lipids to 100 mM in EtOH and allow sufficient time for solubilization.
- Bring DSPC and DMG-PEG-2000 to 10 mM in EtOH and cholesterol to 100 mM in EtOH.
a. These may require heating to 60-65C for complete solubilization in EtOH.
b. Cholesterol in solution should be kept warm (>37C) to maintain solubility. - For each mL of lipid stock add the following:
125 uL of 100 mM ionizable lipid, 250 uL 10 mM DSPC, 96.25 uL 100 mM cholesterol*, 37.5 uL DMG-PEG-2000, and 491.25 uL EtOH.
*Note: cholesterol solution should be transferred as quickly as possible to avoid cooling within any tubing or pipette tips used for preparation.
This should result in 25 mM total lipid mix with 50% ionizable lipid (12.5 mM), 10% DSPC (2.5 mM), 38.5% cholesterol (9.625 mM), 1.5% DMG-PEG-2000 (0.375 mM). All lipid solutions and lipid mixes should be sealed and stored at –20C.
Preparation of RNA-LNPs
This procedure is for a 5:1 RNA:lipid mix (v:v). In our optimization experiments, the GFP mRNA used was diluted to 0.4 uM in sodium acetate buffer prior to mixing with lipids. LNPs may be prepared by hand mixing or by automated microfluidic devices. The procedure below describes small scale preparation using hand mixing which can be scaled as needed.
- Bring lipid mixes to room temperature prior to use and vortex as needed to mix.
- Thaw RNA stocks on ice.
- Once thawed, bring the RNA to the desired concentration using sodium acetate buffer.
- Calculate the amount of lipid required to achieve a 5:1 RNA:lipid ratio. Ex: for every 100 uL of RNA add 20 uL of lipid mix.
- Add the lipid mix to the RNA solution and mix rapidly. The solution should become cloudy and opaque. We do not recommend mixing by vortex as this may shear free RNA.
- Once the LNPs have formed, they should be diluted or dialyzed into a neutral pH buffer such as PBS.
For small scale preparations a 20x dilution is sufficient to neutralize pH and dilute out the EtOH from the lipid mix. Ex: LNPs formed in a 5:1 RNA:lipid ratio with a 100 ug/mL concentration can be diluted 20x to 5 ug/mL RNA and then diluted further in buffer, vehicle, or media for testing in vitro or in vivo testing. Dialysis should be used if higher RNA concentrations are desired or if the volume of LNPs prepared is sufficiently large, i.e. >5-10 mL. All LNPs should be 0.2 micron filter sterilized before use if not prepared under sterile conditions.
Components & Cargo
For most LNPs, four types of lipids are required for formation:
Ionizable Lipid: This is the key component of the LNP (35-50%) which binds and releases the RNA or other cargo in the cell. Examples: ALC-0315, cKK-E12, SM-102, and Dlin-MC3-DMA
PEGylated Lipid: Small amounts of a PEG derivatized lipid (0.5-3%) is incorporated to increase the circulatory half-life in the body. Examples: ALC-0159, DSPE-mPEG, and DMG-mPEG
Cholesterol: Cholesterol is a structural “helper” lipid that makes up a significant part of the LNP (40-50%) and improves efficacy possibly by promoting membrane fusion and promoting endosomal escape.
Neutral phospholipids: Synthetic phospholipids (~10%) are also commonly used as structural “helper” lipids in LNP formulation to promote cell binding.

Technical Notes
- Dried lipids resuspended in ethanol may require additional dilution or heating for complete solubilization as outlined in the assay protocol.
- The assay protocol provides steps for preparing and mixing lipids in ethanol. Alternative organic solvents were not tested and should be evaluated independently by the user.
- RNA stocks should be prepared at sufficiently high concentrations prior to dilution with the RNA Dilution Buffer. In our optimization experiments, RNA was stored at -80°C at 1 mg/mL in aqueous buffer.
- RNA-LNPs diluted or dialyzed to neutral buffers are stable for at least 30 days at 4°C. Longer term storage at -20°C is possible but some loss of activity may occur due to freeze-thaw effects on the lipids. Addition of cryoprotectants or lyophilization of RNA-LNPs for storage at -20° to -80°C should be evaluated by the user.
- Successful delivery of RNA cargo may depend on cell type and the ionizable lipid being used. Results may also vary depending on the type of cell culture system, i.e. monolayers vs. 3D cultures.
- Use of serum free media for application of RNA-LNPs in cell culture is not required.
Testing & Validation
For our optimization tests we paired an enhanced green fluorescent protein (EGFP) mRNA with different LNP formulations. Molar ratios of the four lipid components were kept constant with the ionizable lipid varying across test conditions.
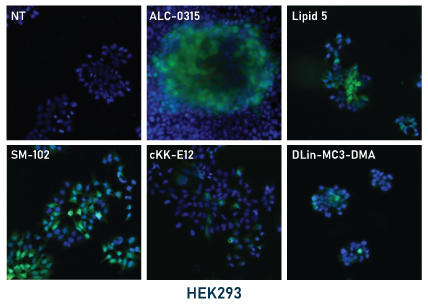
In our experiments, the mRNA-LNPs were prepared by hand mixing using pipettes. LNPs were diluted to 5 μg/mL RNA in PBS and stored at 4°C for the duration of the experiments. For application to cell cultures, LNPs in PBS were diluted 10-fold to 0.5 μg/mL in culture media and then applied directly to the cells. Cells were then monitored by fluorescence microscopy for GFP expression then fixed and mounted for fluorescence imaging (shown below, left panel). GFP expression was apparent for all tested ionizable lipids within 48 hours with some conditions showing expression within 24 hours. Expression was stable up to 96 hours.
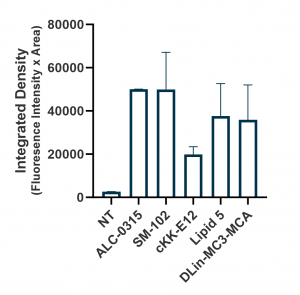
Relative expression levels were quantified and plotted using ImageJ and GraphPad (below, right panel). While GFP fluorescence was observed with all tested ionizable lipids, we observed some variation depending on cell density and growth orientation, i.e. 2D vs 3D. This was indicative of some ionizable lipids being more efficacious in a 3D or tissue-like environment.
For characterization of LNPs, we primarily focused on encapsulation efficiency. Brief protocols are described below:
Quantitative – fluorescence-based quantification using reagents such as QuantiFluor®, Quant-it RiboGreen™, or similar
products.
- Prepare detection reagents as described by manufacturer in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8.0).
- Prepare a 1% v/v solution of Triton X-100 in TE buffer.
- Add the 1% Triton X-100 solution to an aliquot of the LNPs for a 0.2% v/v final concentration and incubate on ice for 10-15 minutes. Add an equivalent volume of TE alone to separate aliquots of the LNPs.
- Add the prepared detection reagent to a 96-well plate (white or black, not clear) and add the required amount of LNP samples (+Triton or +TE). Mix briefly and incubate at room temperature for 5 minutes. Protect from light.
- Read the plate according to the excitation/emission spectra for the detection reagent.
- Encapsulation efficiency (EE) is calculated as follows:
EE = 100 * (T-U)/T
where ‘T’ is the total RNA in the sample (+Triton) and ‘U’ is the free, unencapsulated RNA (+TE). For absolute quantification of total encapsulated RNA, an RNA standard with known concentration should be run in parallel with the samples.
Qualitative – agarose gel-based assessment of encapsulated/protected RNA.
- Prepare a 1% agarose (w/v) TAE gel with sufficient lanes for all samples and a gel ladder. Add nucleic acid dye such as SYBR Safe at a 1:10,000 dilution.
- Prepare a 1% v/v solution of Triton X-100 in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 8.0).
- Add the 1% Triton X-100 solution to an aliquot of the LNPs for a 0.2% v/v final concentration and incubate on ice for 10-15 minutes. Add an equivalent volume of TE alone to separate aliquots of the LNPs.
- Add gel loading buffer to LNP samples (+Triton or +TE) and load all samples and the gel ladder to the prepared gel.
- Run the gel for approximately 45 minutes at 100V and image under UV light.
Successful LNP preparation and encapsulation should result in little to no visible bands for the lanes of the gel with +TE (untreated) samples as the RNA is protected from the dye in the gel by the lipids. Conversely, +Triton (treated) samples should have readily apparent bands on the gel due to release of the RNA from the LNPs due to Triton treatment.
0.2
/ 0.2