Back to blog

Previously, we outlined the general feasibility of lipid immunostaining (immunocytochemistry, ICC and immunohistochemistry, IHC) as this technique has historically been thought of as protein specific. Considering the increasing interest in lipid detection and quantification, this article will delve into specific aspects of lipid immunostaining that should be considered for a successful experiment.
Fixation
Chemical fixation is considered a general requirement for immunostaining with antibodies outside of a live-cell context. By far, the most common reagents used for fixation of cells are formaldehyde (FA) and methanol. However, these reagents achieve fixation in very different ways.
FA is a well-studied crosslinking reagent that reacts readily with free amines; thus, it easily reacts with the N-terminus of proteins or amino acids that have amine groups1. Methanol on the other hand is an organic solvent which precipitates proteins by dehydration rather than crosslinking them. Methanol, and similar solvents like acetone, may be preferable for protein staining due to reductions in background signal, but these can solubilize and remove lipids from cells. For this reason, we generally do not recommend the use of methanol or similar solvents for fixation. If solvent fixation is unavoidable due to the need to co-stain a protein under these conditions, then we recommend comparing the staining results with a crosslinker and solvent for the lipid of interest.
An additional parameter of fixation to consider is the stringency of the crosslinking reaction. For instance, the concentration of FA may be varied as a means of shifting the extent to which cells and proteins are fixed. Other common reagents such as glutaraldehyde (GA) may also be added to facilitate fixation. GA reacts with similar chemistry as FA, but due to its longer carbon chain the proximity of the crosslink it produces will not be as tight as that observed with formaldehyde. The effect of stringency can be seen in Figure 1 where an antibody to phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) has been employed.
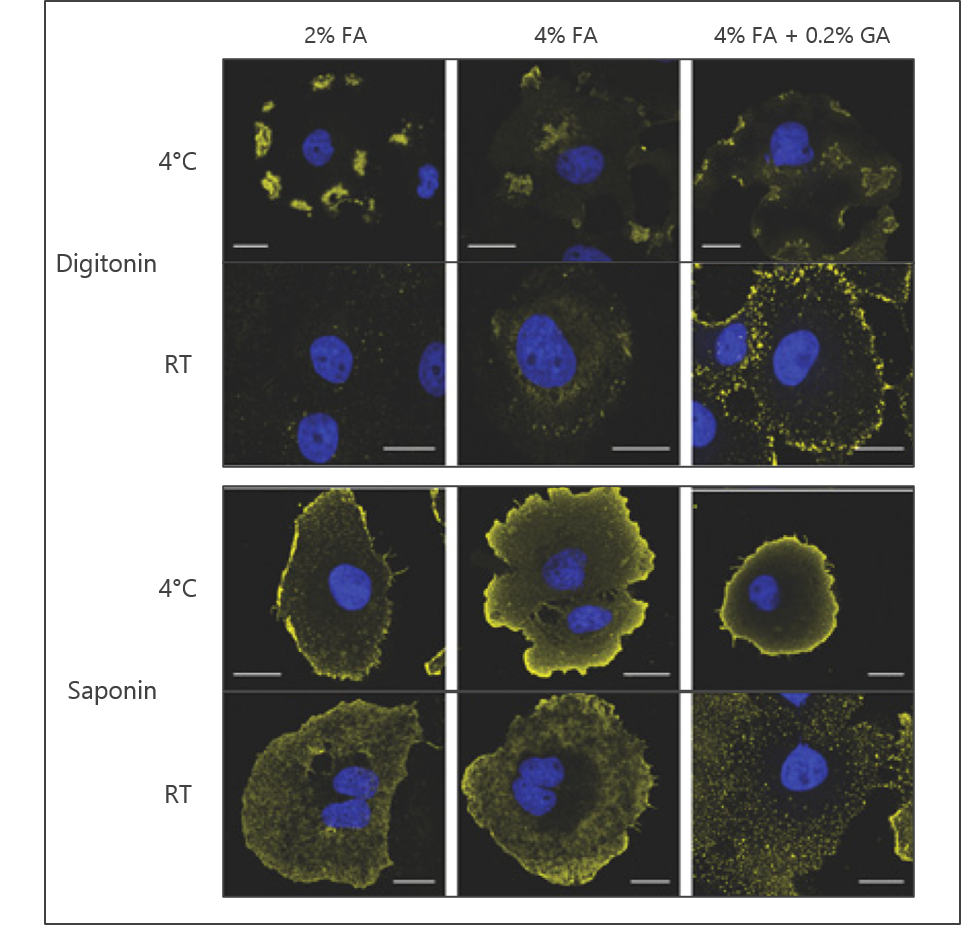
Figure 1: The effects of fixation stringency, permeabilization reagent, and staining temperature on the observed signal using a lipid antibody. Representative signal from anti-PI(4,5)P2 antibody is shown in yellow; nuclear DNA staining with DAPI is shown in blue. Images adapted from Hammond et al. 2009.
Permeabilization
Permeabilization in this context typically refers to the usage of some type of detergent to create holes or pores in the cell membrane to allow access for the staining antibodies. The detergent used here can be highly flexible for proteins given the primary goal of accessing the intracellular compartment. For lipids however, more care must be taken in choosing a permeabilization reagent as they have different solubilizing effects on specific cellular compartments.
The most used reagent for permeabilization is Triton X-100. This non-ionic detergent is non-specific and will generally permeabilize all cellular membranes, however it may completely solubilize some structures like the Golgi apparatus or the endoplasmic reticulum. At sufficiently high concentrations it can also extract proteins or lyse cells which can either affect signal localization or overall intensity. Conversely, it is the only common permeabilization reagent that can effectively penetrate the nuclear membrane.
Saponin and digitonin are also commonly used for permeabilization (Figure 1). Saponin is amphipathic and digitonin is non-ionic, but both are selective for cholesterol. This selectivity allows for efficient permeabilization of the plasma membrane but not for some other organelles like the nucleus or mitochondria. These detergents are also largely reversible in that they can wash out if not maintained during the staining procedure.
Temperature
With many antibody-based detection methods, incubation steps can be readily interchanged between room temperature (RT) and 4°C without meaningful changes to the results. However, for ICC with lipids temperature must be given more consideration.
As mentioned above, FA and GA are the most common reagents used for fixation of cells and mostly react with primary amine groups meaning that they will chiefly crosslink proteins (Figure 2). While it is possible for these reagents to react with unsaturated acyl chains or with lipids containing amine head groups, it is generally unlikely that FA or GA crosslinking will occur directly with lipids. This means that some lipid movement within cellular membranes is likely to be maintained even with fixation.
The extent of movement of a lipid of interest post-fixation will be predominantly affected by two things: the relative fluidity of the specific cellular membrane and the temperature the at which the staining is conducted (Figure 1). For these reasons, it may be advisable to perform the staining procedure at 4°C if the target cellular membrane is known to be highly fluid. Post-fixation of the cells after the secondary antibody is applied can also reduce potential lipid movement as the affixed antibody can be crosslinked to neighboring proteins.
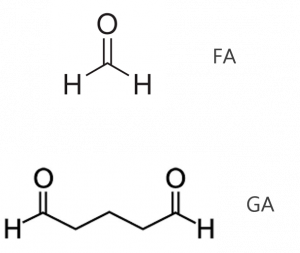
Figure 2: Chemical structures of formaldehyde and glutaraldehyde, two common cellular fixatives.
Controls
As with any antibody-based method, a primary concern is the specificity of the observed signal. Common techniques for addressing specificity for proteins include the use of knockout animals or cell lines as well as preincubation with blocking peptides.
Preincubation and other competitive techniques are more easily adapted for use as controls with lipid antibodies. One simple approach that we often recommend is to preincubate the primary antibody with an array of liposomes carrying various lipids. An example of this can be seen in Figure 3 where an antibody to PI4P has been preincubated with liposomes loaded with various phosphoinositides. As seen in the figure, loss of signal is observed only when PI4P liposomes are applied indicating that the signal is indeed specific. The use of free, water-soluble diC8 lipids may also provide similar results but this will vary between antibodies and should be determined empirically.
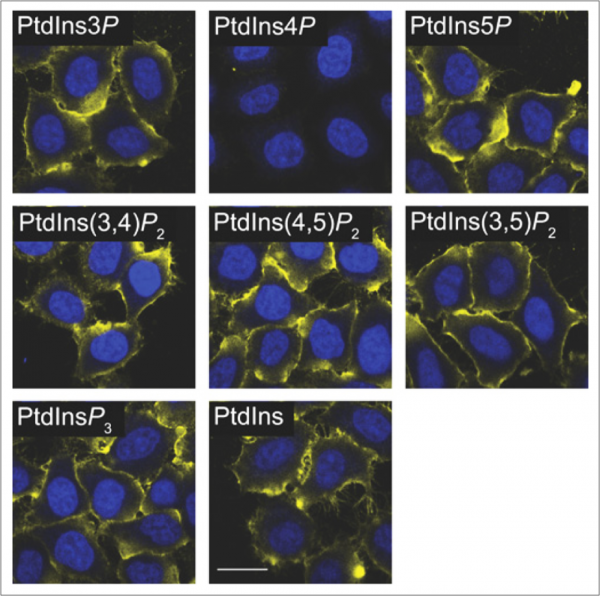
Figure 3: An anti-PI4P antibody was preincubated with various PIPn liposomes as a control for antibody staining. Loss of signal is only observed with PI4P liposomes. Images adapted from Hammond et al. 2009.
An alternative to liposome preincubation is the inclusion of a lipid binding domain where one is available and specific. Figure 4 shows plasma membrane staining with a PI(4,5)P2 antibody (Fig. 4a). In panels b and c, the cells are preincubated with lipid binding domains for either PI(4,5)P2 (PH-PLCδ) or PI(3,4,5)P3 (PH-GRP). Antibody staining is lost only when the PI(4,5)P2 binding domain is applied, again indicating signal specificity.
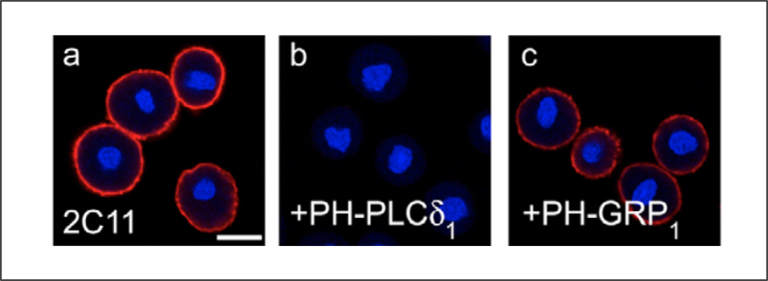
Figure 4: Cells were pretreated with either a PI(4,5)P2 binding domain (b) or PI(3,4,5)P3 binding domain (c) were compared to anti-PI(4,5)P2 treatment alone (a) Loss of signal is only observed with application of the PI(4,5)P2 binding domain. Images adapted from Hammond et al. 2006.
Additional considerations
Questions regarding quantification of lipid levels based on antibody staining are also common. Given the possible differences in staining patterns that can arise from varying fixation and permeabilization techniques, it is unlikely that a single staining method will reveal the entire cellular level of a given lipid. Additionally, endogenous lipid binding proteins are likely to remain bound to their target lipids after fixation and can mask some of the available lipid pools. While these points do not prohibit comparison of signal intensity between different treatment conditions, it is probably wise to think of the resulting data as accurate, but relative quantification.
Alternatives to the controls described above are also possible, but great care should be applied in their application. The use of agonists, inhibitors, or genetic manipulations that are known or expected to produce an increase or decrease for a given lipid can be used as controls. The timing, duration, and/or concentration of these reagents can greatly affect the response of their attendant signaling pathways. For this reason, an observed deviation from the expected result does not equate to non-specificity of an antibody. If unexpected results from a specific stimulus are observed post staining then it is generally advisable to revisit the experiment with the competitive, preincubation controls described above.
Summary
The myriad issues that can arise during lipid immunostaining can seem overwhelming. However, this should not deter one from the use of lipid antibodies in these contexts. Many lipid antibodies are already well characterized and reliable, representative results can be found in the scientific literature. The points raised in this article are intended as a guide and are most useful where an established lab protocol is not available, or a new experimental system is being tested.
Our recommended starting protocols based on the points outlined above can be found here. References for various antibodies and other assay notes are also included. Happy staining!
References
- Guo Z., Chunfang T., Yanrui Y., Liu K. Immunofluorescence staining of phosphoinositides in primary mouse hippocampal neurons in dissociated culture. STAR Protocols 2022 (3)
- Elizabeth A. Hoffman, Brian L. Frey, Lloyd M. Smith, and David T. Auble Formaldehyde Crosslinking: A Tool for the Study of Chromatin Complexes (2015) JBC 290, NO. 44, pp. 26404 –26411
- Hammond GR, Dove SK, Nicol A, Pinxteren JA, Zicha D, Schiavo G. Elimination of plasma membrane phosphatidylinositol (4,5)-bisphosphate is required for exocytosis from mast cells. J Cell Sci. 2006;119(Pt 10):2084-94.
- Hammond GR, Schiavo G, Irvine RF. Immunocytochemical techniques reveal multiple, distinct cellular pools of PtdIns4P and PtdIns(4,5)P(2). Biochem J. 2009;422(1):23-35.
0.2
/ 0.3
Related Articles
Stay informed with our informative blog posts.

Inhibitor Screening with a Hyaluronidase Activity ELISA
Hyaluronic acid (HA) is a fascinating bioactive polymer involved in various cellular signaling pathways. It has been extensively s

Lipid Nanoparticles: Formulation Concepts + Guidance
The advent of the vaccines for SARS-COV-2 / COVID19 has generated an explosion in interest in creating new medicines based on thei

Intracellular Delivery of Phosphoinositides: Expanded Applications
In the realm of biological research, the efficient delivery of molecules into cells is a fundamental yet often challenging task. S

How specific is the LPA Assay Kit II?
In this blog post, we will present the specificity data for the Lysophosphatidic Acid (LPA) Assay Kit II (cat# K-2800S). This ELIS
Why care about PI4-Kinase?
PI4-kinase, or phosphatidylinositol 4-kinase (PI4K), is an enzyme involved in the synthesis of phosphatidylinositol 4-phosphate (P
Lipid Nanoparticles for RNA Delivery
Genetic therapies have now been pursued for several decades and close to a dozen DNA-based gene therapies have gained regulatory a
0.3
/ 0.3
Get in Touch
If you have any questions or would like to learn more about our services, feel free to reach out to us. We’re here to help!
Biosciences